This content is part of a ‘Black British Women on Health’ series of articles in partnership with Wellcome Collection.
In my Year 9 netball team, my position was wing attack. Rounders season in the late spring was the highlight of my school year. I was positioned second in the 400-metre relay for sports day. Competitive sports lit me up, so I pushed myself to get better.
But my determination was met with disappointment after often letting down the team because I was breathless and hunched over in pain.
It took multiple GP appointments over five years to be taken seriously. It baffled me why I had sharp pains under my left rib while running or even just keeping pace with whoever I was walking with, yet no one else did.
I still recall waiting in paediatric outpatients with my dad, in my school uniform, without the faintest idea of what was wrong with me. Neither of us did.
At age 14, I received my fate while my year group chose their GCSE options. I was diagnosed with sickle cell anaemia HbSS, the most severe kind of sickle cell disease (SCD). The first thing that came to mind was: is this the end?
It wasn’t the end, but the beginning of a lifelong, complicated health journey due to poor luck in the genetic lottery. If you or your partner carry the gene, there is a one in four chance of conceiving a child with the condition. It’s usually detected during pregnancy or soon after birth through blood-sample screening. Still, somehow, I slipped through the net.
Sickle cell disease is the world’s most common genetic blood disorder and the fastest-growing in the UK. It is estimated that around 15,000 people live with it. In SCD, red blood cells are inflexible and crescent-shaped, inhibiting blood flow. Sickled red blood cells die more quickly than normal red blood cells, reducing oxygen transported around the body. This leads to fatigue and shortness of breath.
False beliefs about Black people’s pain
I've had soul-crushing pain crises caused by sickled blood clogging up my blood vessels. In those moments, I've seen the light! Sometimes it was the morphine administered to ease the pain and other times it was while I gripped onto life with every laboured breath.
For having endured suffering like this, I've been awarded the name “sickle cell warrior”. This tokenistic term bandied around the sickle cell community felt vapid, so never formed part of my vocabulary.
The issue with being deemed a “sickle cell warrior” is that it can feed assumptions that we can handle the pain, and then some. Those affected by SCD are predominantly from African, Caribbean, Middle Eastern or Mediterranean heritage.
This tokenistic term bandied around the sickle cell community felt vapid, so never formed part of my vocabulary.
Still, there is a misconception it's a disorder only present in the Black community. This presents further challenges for sicklers, as there is a pervasive subconscious belief woven into the medical community that Black people have thicker skin than white people.
I reject the term “warrior”, but I can’t deny many of us go through a lot every day, often in isolation. My sickle cell journey has been punctuated with many moments of acute pain or complications like pneumonia. Even when you’re not in crisis, it's a disease that never leaves your side. Few witness our rapid decline in health or daily chronic battles.
Stress, sugar and time off sick
SCD followed me through university. Late nights spent drinking mindlessly while wearing short skirts and mesh tops in midwinter led to my first A&E admission for severe pain. Back then, I tried to turn a blind eye to my triggers, many of which are common among sicklers: cold temperatures, extreme change in temperature, dehydration, overexertion, lack of sleep, a poor diet heavy in sugar and low in nutrients, high altitudes, and stress.
Stress in particular became a linchpin, sending me into a cascade of crises and health complications. I can almost get away with being dehydrated or eating a diet rich in Cadbury's chocolate. But paired with low-level chronic or intense, acute stress, these things trigger pulsating pains for days in my back, legs or arms, or sometimes in more than one place.
SCD follows me on dates. While I just want to avoid men that give me the ‘ick’, I often try to avoid falling for men with SCD too. See, I don't want my future children to suffer as I have. So it's best to have them with someone without SCD or carriers of the gene. Opening up early works well for me, so I can gauge their awareness and hopefully learn if they have it too.
SCD follows me to work, too: 10 pm finishes, 8 am starts, after-work drinks, and the pressure to go the extra mile means I've struggled climbing the corporate ladder. Burning the candle at both ends has nearly cost me my life, but even after spending two weeks in the hospital, I was nervous about jeopardising my job.
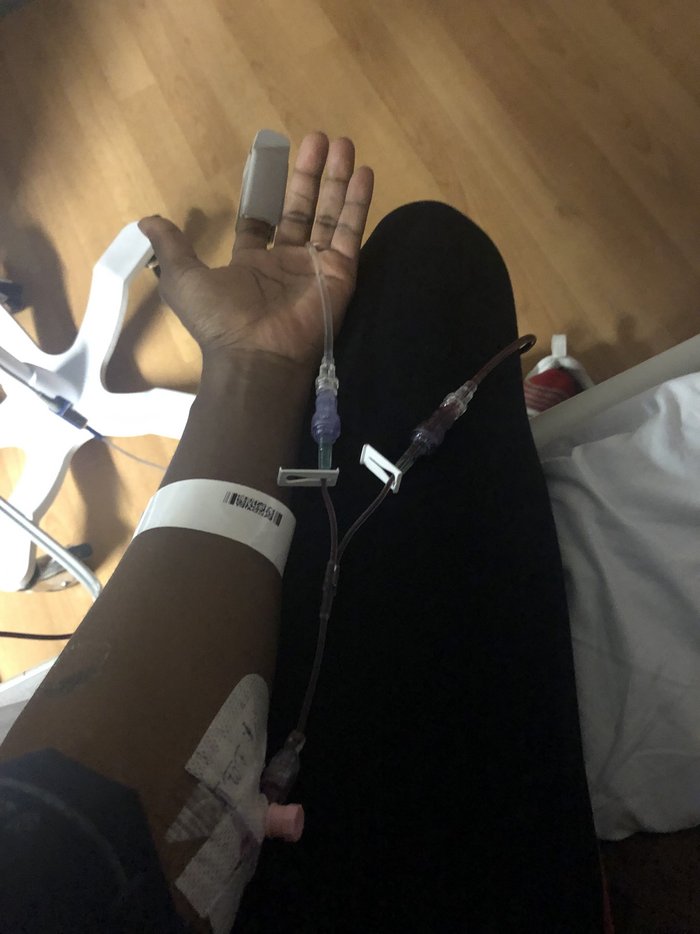
One boss noted my absenteeism was “comparatively high”. Calling in sick most months and taking off half-days for hospital appointments, despite putting in double time when I was clocked in, apparently warranted that. The culture was far from inclusive and stubbornly inflexible, which led to many sick days and, eventually, quitting.
Since then, the corporate world has increasingly focused on employee wellbeing, especially with the introduction of hybrid working, which benefits those with chronic health conditions who need more flexibility.
However, long before work-life balance trended, I had no choice but to prioritise it. I said no to frequent late finishes and fought for the option to work from home before it became the norm. Simple tasks, such as a daily rush-hour commute, drain my energy, stressing me out before I'm at my desk.
Being misunderstood and gaslit
Unless I'm in visible pain, crying, shouting, rocking and struggling to breathe, I look perfectly well to family, friends, bosses, exes and even doctors. I was taken aback when one doctor told me I was “glowing”, despite presenting dangerously low blood-oxygen levels.
The sad truth is that many sicklers feel unheard, and many times gaslit. Stories like Evan Smith's, a 21-year-old man with SCD, who died after being denied proper treatment, are not uncommon. An inquest heard that he desperately called 999 from his North Middlesex hospital bed for oxygen after nurses said he didn't need it. His story hit home: I had also struggled to receive adequate care in the same hospital at age 21.
I'm tired of fighting for care, and I know many others are too. The burden of SCD can no longer be carried by the "warriors". We need allies!
The possibility of a cure is closer than ever, but while one is yet to come to fruition, we need allies to help in a way that can make a huge difference today.

Blood transfusions are a frequent treatment for prolonged pain crises, and I have benefited from them countless times. Transfusions decrease the percentage of sickled red blood cells. This can help treat severe anaemia and reduce the risk of stroke and other complications of blocked blood flow.
Blood donation within the Black, Asian and minority ethnic communities in the UK only accounts for five per cent of all donations.
Receiving blood from a Black or similar ethnicity donor can be more beneficial. The CDC states there needs to be “matching antigens or special proteins on the surface of each red blood cell” to our blood. And rare blood groups like Ro subtype O Rh positive and B positive are more prevalent among Black, Asian and minority ethnic communities.
Sicklers don't need tokenistic accolades. We need blood that will carry us through our careers and on our adventures. Blood that lets us compete in sports or go on brisk walks. Blood that will keep us warm and allow our organs to function as they should. We need blood that raises our oxygen levels and starves the pain. We need your blood!
Header image of Cheryl Telfer in Morocco.
Read more from the Black British women on health series



